This page was generated from /home/docs/checkouts/readthedocs.org/user_builds/asapdiscovery/checkouts/latest/docs/_collections/notebooks/exploring_related_sequences_and_structures.ipynb.
Exploring related sequences and structures
drugforge-spectrum tool. This tool simplifies the design of broad-spectrum antivirals by enabling users to: 1) search for all the related sequences of a protein target of interest, and 2) generate and align the structures of these related targets for further visualization and testing.This tutorial will guide you through the process of querying a protein sequence on BLAST to find related proteins and align their sequences and structures. The notebook is divided as follows:
Runing a BLAST search on a query protein sequence.
Performing a multi-sequence alignment between the reference and a sub-set of the matching sequences, based on a selection criteria.
Preparing a job on ColabFold to generate structures of the related proteins.
Generating a PyMol session with the structures of reference and related proteins.
How to do all the above steps in the command-line!
[2]:
from pathlib import Path
import pandas as pd
from drugforge.spectrum.blast import get_blast_seqs
from drugforge.spectrum.seq_alignment import Alignment, do_MSA
from drugforge.data.testing.test_resources import fetch_test_file # fetch test files
import warnings
warnings.simplefilter("ignore", ResourceWarning)
from Bio import BiopythonDeprecationWarning
warnings.simplefilter("ignore", BiopythonDeprecationWarning)
# Print info logs
import logging
logging.basicConfig(
level=logging.INFO,
format='%(levelname)s: %(message)s',
force=True
)
We will save our results from this run on a separate directory, because we expect a lot of output files!
[3]:
output_dir = "tutorial_results"
output_dir = Path(output_dir)
output_dir.mkdir(parents=True, exist_ok=True)
BLAST search on a query protein sequence
We start with an input fasta file, containing the sequence of the protein of interest. For example, the fasta sars-cov2-mpro.fasta contains the sequence of one of the chains in SARS-CoV-2 Mpro.
[4]:
input_fasta = output_dir/"sars-cov2-mpro.fasta"
with open(input_fasta, "w") as f:
f.write(">SARS-CoV-2 Mpro Chain A, B\n")
f.write(
"SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTSEDMLNPNYEDLLIR" \
"KSNHNFLVQAGNVQLRVIGHSMQNCVLKLKVDTANPKTPKYKFVRIQPGQTFSVLACYNG" \
"SPSGVYQCAMRPNFTIKGSFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGN" \
"FYGPFVDRQTAQAAGTDTTITVNVLAWLYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYE" \
"PLTQDHVDILGPLSAQTGIAVLDMCASLKELLQNGMNGRTILGSALLEDEFTPFDVVRQCSGVTFQ"
)
Bio.Blast.NCBIWWW module from BioPython, which queries from the NCBI BLAST server. There’s a few parameters from BLAST we are interested in, because they control the number of matches we get from our search:nalign: The number of database sequences BLAST will show alignments for.nhits: The number of hit sequences to return.e_val: Expect value (E) for saving hits.
In general, we want to maximize the number of sequence matches! In the next step we will learn how to apply other filters to our BLAST output, but we wold like to have as big of a pool to select from as possible.
We will use the defaults nalign=500 and nhits=100 for this tutorial to limit the size of our output, but we recommend using >1000 for the alignments and a similar number for the hits for production runs (by definition, keep nalign>nhits). The BLAST default for e_val is 10, which is a good value for exploratory searches. You can improve the quality of your search by decreasing the expect value, at the risk of limiting the size of your output.
We will also save the output of the BLAST run to a CSV file for book-keeping.
[27]:
save_csv = "blast_log.csv"
[28]:
matches_df = get_blast_seqs(
input_fasta,
output_dir,
input_type="fasta",
save_csv=save_csv,
nalign=500,
nhits=100,
e_val_thresh=10,
verbose=False,
xml_file="results.xml"
)
matches_df.head()
BLAST search with 500 alignments, expect 10, 100 hitlist_size and 100 descriptions
[28]:
| query | ID | description | sequence | host | organism | score | |
|---|---|---|---|---|---|---|---|
| 0 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009725295.1| | ORF1a polyprotein [Severe acute respiratory sy... | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTS... | 100.00 | ||
| 1 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009724389.1| | ORF1ab polyprotein [Severe acute respiratory s... | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTS... | 100.00 | ||
| 2 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009725301.1| | 3C-like proteinase [Severe acute respiratory s... | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTS... | 100.00 | ||
| 3 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009944365.1| | ORF1a polyprotein [SARS coronavirus Tor2] | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDTVYCPRHVICTA... | 96.08 | ||
| 4 | SARS-CoV-2 Mpro Chain A, B | ref|NP_828849.7| | ORF1ab polyprotein [SARS coronavirus Tor2] | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDTVYCPRHVICTA... | 96.08 |
Alternatively, we could already have a PDB file with our reference’s crystal structure. We can initialize the BLAST search with the PDB file and generate a fasta file from our PDB structure, by changing the input_type to "pdb".
WARNING: This run will take longer than the previous one, because BLAST discourages multiple queries of the same sequence within a short time frame. You can skip this cell if you’d like, the rest of the notebook will run!
[ ]:
input_pdb = fetch_test_file("Mpro-P2660_0A_bound.pdb") # SARS-Cov-2 MPRO structure
'''
matches_df_pdb = get_blast_seqs(
input_pdb,
output_dir,
input_type="pdb",
save_csv=save_csv,
nalign=500,
nhits=100,
e_val_thresh=10,
verbose=False,
xml_file="results_pdb.xml"
)
matches_df_pdb.head()
'''
Aligning the sequences from our BLAST search
Now that we have a list of protein sequencies that are related to our protein of interest, we want to visualize the similarities between the sequences. We will do this by aligning and visualizing multiple sequences from the BLAST output.
Filtering our sequences based on a keyword match
The number of sequences returned by BLAST may get quite large, making it hard to find similarities between the protein sequences. Additionally, we may not even be interested in all of these! Some entries often share the same sequence (for example, diferent chains of the same protein), and others may simply not be relevant to us. For example, we could be interested in viral proteins that infect humans or we could be interested in MERS-CoV variants specifically.
Information about a given sequence record can be found in the description provided by BLAST. The easiest way to filter our sequence is then to search for a specific keyword in this description, such as “human” or “MERS”.
[30]:
matches_df['description']
[30]:
0 ORF1a polyprotein [Severe acute respiratory sy...
1 ORF1ab polyprotein [Severe acute respiratory s...
2 3C-like proteinase [Severe acute respiratory s...
3 ORF1a polyprotein [SARS coronavirus Tor2]
4 ORF1ab polyprotein [SARS coronavirus Tor2]
...
95 ORF1ab polyprotein [Lucheng Rn rat coronavirus]
96 nsp5 [Canada goose coronavirus]
97 ORF 1ab polyprotein [Beluga whale coronavirus ...
98 polyprotein ORF1a [Camel alphacoronavirus]
99 polyprotein ORF1ab [Camel alphacoronavirus]
Name: description, Length: 100, dtype: object
Once we decide the keyword for our filter, we proceed to align sequences based on our filter. For this we define an Alignment object, which will take care of both tasks, given a query sequence (“sars-cov2-mpro”) and the DataFrame we obtained from the BLAST search:
[31]:
query = matches_df["query"][0]
alignment = Alignment(matches_df, query, output_dir)
From this alignment object, we will filter and align the resulting sequences. The MAFFT multi-sequence aligment tool will take care of the latter. Both steps will be run by the do_MSA() function, which takes the Alignment object and the selection parameter. We also set the parameter n_chains=1, because our PDB structure contains only one chain. This parameter is not relevant for the visualization of the sequences (all BLAST entries correspond to a single chain), but will be useful
for the next structure-alignment step of this tutorial.
[33]:
sel_key = "human"
file_prefix = "sars_tutorial"
alignment = do_MSA(
alignment=alignment,
select_mode=sel_key,
file_prefix=file_prefix,
plot_width=2000,
n_chains=2,
color_by_group=True,
start_alignment_idx=1,
max_mismatch=2,
custom_order=""
)
INFO: A fasta file tutorial_results/sars_tutorial.fasta have been generated with the selected sequences
INFO: A fasta file tutorial_results/sars_tutorial_alignment.fasta have been generated with the multi-seq alignment
INFO: A csv file tutorial_results/sars_tutorial.csv have been generated with the selected sequences
INFO: The multi-sequence alignment returns the following matches:
INFO: group: 181/307
INFO: exact: 89/307
INFO: none: 37/307
INFO: Aligning 5 sequences of lenght 307
INFO: A html file tutorial_results/sars_tutorial_alignment.html have been generated with the aligned sequences
This step should be completed pretty fast! You should see the following files in your output_dir:
A
fastafile with the ID, description and sequence of the entries selected via the provided keyword.An
.htmlfile with a colored representation of the multi-sequence alignment (mismatches in red, and group matches up tomax_mismatchesin yellow). The width of the plot can be adjusted viaplot_widthoption.A
fastafile with the aligned sequences (with “-” representing gaps inserted)A CSV file with the ID and sequence of the entries selected via the keyword. We will use this output in the next step of this tutorial.
Sequences can also be selected based on the virus host, using the Entrez database
Entrez is a molecular biology database system that provides integrated access to nucleotide and protein sequence data. Importantly, it provides information on the organism and host of a given viral protein sequence. We are now going to query the Entrez database to filter our sequences based on viral host information.
We retrieve Entrez host information when we process the BLAST search, and the host and organism information will be included in the BLAST CSV output. In the previous call to the get_blast_seqs() function, however, no query was made to the Entrez database. The function will perform the database search only if a valid email address is provided via the email parameter. This email address is required by the Entrez API, and is used as way of controlling how much a single user queries the
database.
We have to re-run the function, but there is not need to run the entire BLAST search from scratch! If you look through your results folder, you will notice a new file results.xml was saved. This file contains a backup of the most recent BLAST search and can be used to retrieve the results from the run. This is really convenient as BLAST will take longer each time we submit the (same) sequence, so after a few queries the run will take 10+ mins.
[ ]:
input_presaved = output_dir / "results.xml"
matches_df = get_blast_seqs(
input_presaved,
output_dir,
input_type="pre-calc",
save_csv=save_csv,
verbose=False,
email="your.email@email.com" # Uncomment with your own email!!
)
matches_df.head()
| query | ID | description | sequence | host | organism | score | |
|---|---|---|---|---|---|---|---|
| 0 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009725295.1| | ORF1a polyprotein [Severe acute respiratory sy... | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTS... | Homo sapiens | Severe acute respiratory syndrome coronavirus 2 | 100.00 |
| 1 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009724389.1| | ORF1ab polyprotein [Severe acute respiratory s... | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTS... | Homo sapiens | Severe acute respiratory syndrome coronavirus 2 | 100.00 |
| 2 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009725301.1| | 3C-like proteinase [Severe acute respiratory s... | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTS... | Homo sapiens | Severe acute respiratory syndrome coronavirus 2 | 100.00 |
| 3 | SARS-CoV-2 Mpro Chain A, B | ref|YP_009944365.1| | ORF1a polyprotein [SARS coronavirus Tor2] | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDTVYCPRHVICTA... | Homo sapiens; patient #2 with severe acute res... | SARS coronavirus Tor2 | 96.08 |
| 4 | SARS-CoV-2 Mpro Chain A, B | ref|NP_828849.7| | ORF1ab polyprotein [SARS coronavirus Tor2] | SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDTVYCPRHVICTA... | Homo sapiens; patient #2 with severe acute res... | SARS coronavirus Tor2 | 96.08 |
To select based on Entrez information we will use a specific format:
host: <host-species-name> OR organism: <organism-common-name>
host: Homo sapiens OR organism: human is a valid sel_key input that will look for sequences that infect humans by making sure either the corresponding host or organism entry contain the human keywords. It is important to specify both because sometimes Entrez entries do not contain host information but will specify the organism.[ ]:
query = matches_df["query"][0]
alignment = Alignment(matches_df, query, output_dir)
sel_key = "host: Homo sapiens OR organism: human"
file_prefix = "sars_tutorial_human_host"
alignment = do_MSA(
alignment=alignment,
select_mode=sel_key,
file_prefix=file_prefix,
plot_width=2000,
n_chains=2,
color_by_group=True,
start_alignment_idx=1,
max_mismatch=2,
custom_order=""
)
INFO: A fasta file tutorial_results/sars_tutorial_human_host.fasta have been generated with the selected sequences
INFO: A fasta file tutorial_results/sars_tutorial_human_host_alignment.fasta have been generated with the multi-seq alignment
INFO: A csv file tutorial_results/sars_tutorial_human_host.csv have been generated with the selected sequences
INFO: The multi-sequence alignment returns the following matches:
INFO: group: 128/310
INFO: exact: 86/310
INFO: none: 96/310
INFO: Aligning 7 sequences of lenght 310
INFO: A html file tutorial_results/sars_tutorial_human_host_alignment.html have been generated with the aligned sequences
Using ColabFold to generate PDB structures of our sequence matches
So far we have went from a reference protein sequence, to find all related viral proteins that infect humans, and visualizing how much the sequences relate to each other. To analyze how these differences translate into structural differences, we will feed the sequences into ColabFold to generate their corresponding structures with respect to a common template (in this case, the initial reference protein). Then, we will align and visualize the generated sequences with the reference and save a PyMOL session that will allow us to point to specific structural differences.
colabfold_batch program takes as input a CSV file containing the ID and sequence of the structures we wish to fold, as well as the path to the directory all results will be stored.The fold model can also be specified via --model-type. For monomers, “alphafold2_ptm” is often appropiate, while multimers (n_chains>1 in the previous step) can be folded using the “alphafold2_multimer_v3” model. We will run a job with 3 recycles, a single repetition and a monomer model.
The following is an example bash script for running ColabFold with the CSV file generated in the previous step, and the reference PDB saved in the directory template_dir/:
csv_fn="<output_dir>/sars_tutorial_human_host.csv"
template_dir="template_dir" # Ref pdb must be saved here
out_dir="sars_cf_results"
colabfold_batch $csv_fn $out_dir \
--templates --custom-template-path $template_dir \
--num-recycle 3 --num-models 1 --model-type "alphafold2_multimer_v3"
Saving the aligned reference and related sequences on a PyMol session
The following section assumes you have completed the run on ColabFold and have generated structures for all the sequences saved in the <file_prefix>.csv file, created previously.
We will have all the files generated by ColabFold in a folder, for each molecule in the CSV file. If you chose to generate multiple repetitions of each protein fold (via the -random-seed option), you should have multiple PDB files per sequence, all following the same format: ``”<mol_id>_unrelaxed_rank_001_<model_type>_model_1_seed_<seed_number>.pdb”``, where a <seed_number> is assigned to each repetition and <model_type> is the model used for the fold.
Let’s define a function that will go through each <mol_id>*.pdb file in the directory, align and calculate the RMSD of each with respect to the reference and save a PyMOL session of the reference and folded proteins. If we have multiple seed repetitions saved, only those with the least RMSD with respect to the reference will be saved.
[5]:
from drugforge.spectrum.calculate_rmsd import (
save_alignment_pymol,
select_best_colabfold,
)
def gen_alignment_vis(sequence_df, cf_folder, ref_pdb, save_dir, pymol_save):
aligned_pdbs = []
seq_labels = []
for index, row in sequence_df.iterrows():
# iterate over each csv entry
mol = row["id"]
results = Path(cf_folder)
final_pdb = save_dir / f"{mol}_aligned.pdb"
# The output file format in ColabFold depends on the folding method used.
# Here the PDB file format is defined assuming the AlphaFold2_ptm model
# Select best seed repetition
min_rmsd, min_file = select_best_colabfold(
results_dir=results,
seq_name=mol,
pdb_ref=input_pdb, # reference pdb
chain="A",
final_pdb=final_pdb,
fold_model="alphafold2_multimer_v3",
)
aligned_pdbs.append(min_file)
seq_labels.append(mol)
session_save = save_dir / pymol_save
# Save the alignment in a PyMOL session. Here we display only the chain "A", but both can be displayed by setting align_chain="both"
save_alignment_pymol(
aligned_pdbs, seq_labels, ref_pdb, session_save, align_chain="A", color_by_rmsd=True
)
return
If you followed the previous guide, all of the ColabFold results should be saved on the folder sars_cf_results/. For this tutorial, we will fetch previously prepared ColabFold PDBs, but feel free to use your own. Let’s also use the PDB for SARS-CoV-2 introduced in the beginning of the notebook as reference.
[10]:
# grab cf_results
def all_cfold_dir_fns():
return [
"spectrum_dir/YP_009725295_1_unrelaxed_rank_001_alphafold2_multimer_v3_model_1_seed_000.pdb",
"spectrum_dir/YP_009047217_1_unrelaxed_rank_001_alphafold2_multimer_v3_model_1_seed_000.pdb",
"spectrum_dir/YP_009555250_1_unrelaxed_rank_001_alphafold2_multimer_v3_model_1_seed_000.pdb",
"spectrum_dir/NP_835346_1_unrelaxed_rank_001_alphafold2_multimer_v3_model_1_seed_000.pdb",
"spectrum_dir/YP_009944365_1_unrelaxed_rank_001_alphafold2_multimer_v3_model_1_seed_000.pdb",
"spectrum_dir/YP_009944273_1_unrelaxed_rank_001_alphafold2_multimer_v3_model_1_seed_000.pdb"
]
def cfold_dir():
all_paths = [fetch_test_file(f) for f in all_cfold_dir_fns()]
return all_paths[0].parent, all_paths
sars_cf_results, _ = cfold_dir()
[11]:
# grab a reference SARS-CoV-2
input_pdb = fetch_test_file("Mpro-P2660_0A_bound.pdb")
save_dir = output_dir / "aligned_sars_tutorial/"
save_dir.mkdir(parents=True, exist_ok=True)
csv_file = output_dir / "sars_tutorial_human_host.csv"
cf_folder = sars_cf_results
seq_df = pd.read_csv(csv_file)
gen_alignment_vis(seq_df, cf_folder, input_pdb, save_dir, "aligned_proteins.pse")
INFO: RMSD for seed 000 is 1.9166725274687486 A
INFO: YP_009725295_1 seed with least RMSD is 000 with RMSD 1.9166725274687486 A
INFO: RMSD for seed 000 is 3.1352285647682567 A
INFO: YP_009944365_1 seed with least RMSD is 000 with RMSD 3.1352285647682567 A
INFO: RMSD for seed 000 is 2.5524218376956935 A
INFO: YP_009047217_1 seed with least RMSD is 000 with RMSD 2.5524218376956935 A
INFO: RMSD for seed 000 is 2.4124092362555603 A
INFO: YP_009944273_1 seed with least RMSD is 000 with RMSD 2.4124092362555603 A
INFO: RMSD for seed 000 is 2.0554156780668813 A
INFO: YP_009555250_1 seed with least RMSD is 000 with RMSD 2.0554156780668813 A
INFO: RMSD for seed 000 is 2.2915132888118075 A
INFO: NP_835346_1 seed with least RMSD is 000 with RMSD 2.2915132888118075 A
WARNING: No ColabFold entry for YP_010229075_1 and model alphafold2_multimer_v3 found.
After running the above block, the aligned proteins and the PyMOL session will be saved in the folder aligned_ColabFold/. Let’s look at the results, which will show the aligned first chains (chain A), colored by RMSD with respect to the reference protein.
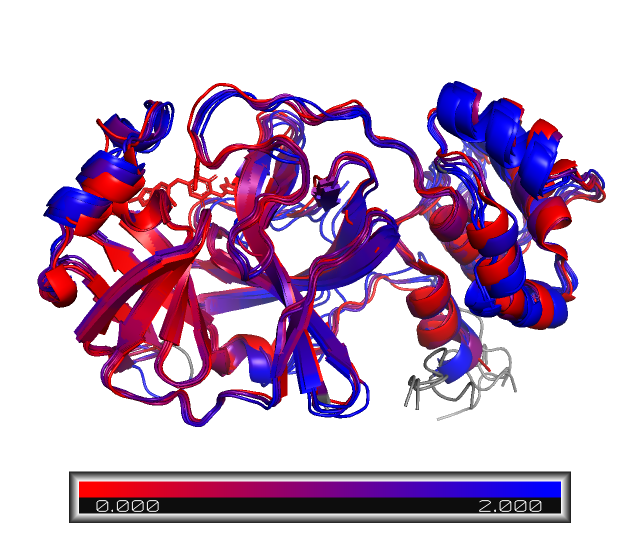
We can clearly see a few differences in the protein structure! These regions are colored in blue. For example, NP_073550_1 and YP_010229075_1, Human coronavirus 229E and NL63, repectively, have a longer aminoacid sequence, as we saw in the sequence alignment step. However, most of the structure is still conserved, especially on the binding site, which is mostly on red.
Scoring ligand affinity in protein-ligand complexes
This tutorial will now explore the protein-ligand scoring features of the spectrum package. As an example, we are going to score the score different properties of a SARS-CoV-2-Ensitrelvir complex, obtained by querying a SARS-Cov-2 sequence and folding (according to the previous steps), and docking and Ensitrelvir ligand. We will determine the strength of interaction based on 3 scores:
A ligand binding affinity estimated with ChemGauss4 (empirical model)
The RMSD with respect to the SARS-CoV-2 reference
A ligand binding affinity estimated with AutoDock Vina (empirical model). [This calculation requires installation of other packages, and it’s only possible on a Ubuntu system]
The protein-ligand interaction fingerprints (COMING SOON)
ChemGauss score
[13]:
from drugforge.spectrum.score import dock_and_score
from drugforge.docking.scorer import ChemGauss4Scorer
logging.basicConfig(
level=logging.ERROR,
format='%(levelname)s: %(message)s',
force=True
)
warnings.filterwarnings("ignore", category=UserWarning)
input_pdb = fetch_test_file("Mpro-P2660_0A_bound.pdb") # Reference PDB for alignment
scorers = [ChemGauss4Scorer()]
comp_name = "test"
target = "SARS-CoV-2-Mpro"
mers_complex = fetch_test_file("SARS-CoV-2_ensitrelvir_folded.pdb")
scores_df, prepped_cmp, ligand_pose, aligned = dock_and_score(
mers_complex,
comp_name=comp_name,
target_name=target,
scorers=scorers,
label=Path(mers_complex).stem,
allow_clashes=True,
pdb_ref=input_pdb,
aligned_folder=output_dir,
align_chain="1",
align_chain_ref="A",
)
print(f"The ligand in {mers_complex.stem} was docked with a confidence of {scores_df['docking-confidence-POSIT'].values[0]}. The ChemGauss score is {scores_df['docking-score-POSIT'].values[0]} kcal/mol.")
scores_df = scores_df[['ligand_id', 'docking-structure-POSIT', 'docking-confidence-POSIT', 'docking-score-POSIT']]
scores_df.head()
No BioAssembly transforms found, using input molecule as biounit: _chain1._chain2_UNK
Processing BU # 1 with title: _chain1._chain2_UNK, chains 123
The ligand in SARS-CoV-2_ensitrelvir_folded was docked with a confidence of 0.99. The ChemGauss score is -9.96858024597168 kcal/mol.
[13]:
| score_type | ligand_id | docking-structure-POSIT | docking-confidence-POSIT | docking-score-POSIT |
|---|---|---|---|---|
| 0 | test | SARS-CoV-2-Mpro | 0.99 | -9.96858 |
Ligand and binding site RMSD with respect to the reference PDB (as implemented in the OpenEye tools)
[32]:
from drugforge.spectrum.score import get_ligand_rmsd
from drugforge.spectrum.calculate_rmsd import get_binding_site_rmsd
from Bio.PDB.PDBExceptions import PDBConstructionWarning
warnings.filterwarnings("ignore", category=PDBConstructionWarning)
lig_rmsd = get_ligand_rmsd(
str(aligned), str(input_pdb), True, rmsd_mode="oechem",
)
df_print = scores_df.copy()
df_print.loc[0,"Lig-RMSD"] = lig_rmsd
pout = f"Calculated RMSD of POSIT ligand pose = {lig_rmsd} A.\n"
bsite_rmsd = get_binding_site_rmsd(
aligned,
input_pdb,
bsite_dist=5,
ligres="LIG",
chain_mob="1",
chain_ref="A",
rmsd_mode="heavy",
aligned_temp=aligned,
)
df_print.loc[0,"Pocket-RMSD"] = bsite_rmsd
pout += f"\nCalculated RMSD of binding site = {bsite_rmsd} A, with ref {input_pdb.stem}"
print(pout)
WARNING: Ref(53) and target(54) ligands have different number of atoms
INFO: The attribute(s) types have already been read from the topology file. The guesser will only guess empty values for this attribute, if any exists. To overwrite it by completely guessed values, you can pass the attribute to the force_guess parameter instead of the to_guess one
INFO: There is no empty types values. Guesser did not guess any new values for types attribute
INFO: attribute masses has been guessed successfully.
INFO: The attribute(s) types have already been read from the topology file. The guesser will only guess empty values for this attribute, if any exists. To overwrite it by completely guessed values, you can pass the attribute to the force_guess parameter instead of the to_guess one
INFO: There is no empty types values. Guesser did not guess any new values for types attribute
INFO: attribute masses has been guessed successfully.
INFO: The attribute(s) types have already been read from the topology file. The guesser will only guess empty values for this attribute, if any exists. To overwrite it by completely guessed values, you can pass the attribute to the force_guess parameter instead of the to_guess one
INFO: There is no empty types values. Guesser did not guess any new values for types attribute
INFO: attribute masses has been guessed successfully.
Calculated RMSD of POSIT ligand pose = 6.794498029514078 A.
Calculated RMSD of binding site = 3.3841235637664795 A, with ref Mpro-P2660_0A_bound
AutoDock Vina affinity
This part of the tutorial requires the installation of the ADFR suite package, which is only available on Ubutu and older versions of MacOS. This is not required by AutoDock Vina per-se, but to convert to the input types it requires for the protein and ligand (.pdbqt). If you have a better way of obtaining those files, you can provide your own path to the prepared files instead.
[33]:
from drugforge.spectrum.score import score_autodock_vina
warnings.filterwarnings("ignore", category=UserWarning)
# Save prepped files for Vina
pdb_prep = output_dir / (aligned.stem + "_target.pdb")
sdf_ligand = output_dir / (aligned.stem + "_ligand.sdf")
prepped_cmp.target.to_pdb_file(pdb_prep)
ligand_pose.to_sdf(sdf_ligand)
# If available, the protein and ligands can be given on pdbqt format, which should work with the current installation
pdb_prep = fetch_test_file("SARS_model_target_prepped.pdbqt")
sdf_ligand = fetch_test_file("SARS_model_ligand_prepped.pdbqt")
vina_box = [-22, 6, 25]
df_vina, out_pose = score_autodock_vina(
pdb_prep,
sdf_ligand,
vina_box,
box_size=[20, 20, 20],
dock=False, # There is an option to dock with Vina, but we will not use it here,
)
df_print = pd.concat([df_print, df_vina], axis=1, join="inner")
INFO: Prepped target provided
INFO: Prepped ligand provided
INFO: Score before minimization: -9.197 (kcal/mol)
INFO: Score after minimization: -9.892 (kcal/mol)
Computing Vina grid ... done.
Performing local search ... done.
We stored all the scores in the df_print DataFrame. There is also an implementation for Gnina scoring (CLI-based) which facilitates the processing of the results, but it won’t be done on this tutorial, as it requires a working Gnina installation.
[34]:
df_print.head()
[34]:
| ligand_id | docking-structure-POSIT | docking-confidence-POSIT | docking-score-POSIT | Lig-RMSD | Pocket-RMSD | Vina-score-premin | Vina-score-min | |
|---|---|---|---|---|---|---|---|---|
| 0 | test | SARS-CoV-2-Mpro | 0.99 | -9.96858 | 6.794498 | 3.384124 | -9.197 | -9.892 |
How to run the sequence/structure alignment in the command-line
The sequence and structure alignment pipelines we went through in the previous steps can also be run from the command line. The drugforge-spectrum CLI includes both pipelines: seq-alignment and struct-alignment, respectively. We will go through the usage and a few examples for each option, using the test case adopted in the tutorial above (SARS-CoV-2 Mpro Chain A).
Sequence alignment usage
Starting from an input protein seqence file, e.g., .fasta, .pdb or pre-computed .xml file, the seq-alignment command will generate all of the alignment files for the target protein and related protein sequences.
The drugforge-spectrum seq-alignment CLI takes as input the sequence file and the path to the folder where results will be stored, along with a series of options to control the BLAST search and alignment outputs. Below is a list of these options, which can also be accessed via the drugforge-spectrum seq-alignment --help option:
-f, --seq-file FILE File containing reference sequences
-t, --seq_type [fasta|pdb|pre-calc]
Type of input from which the sequence will be read. [default: fasta]
--output-dir DIRECTORY The directory to output results to.
--nalign INTEGER Number of alignments that BLAST search will output.
--e-thr FLOAT Threshold to select BLAST results.
--save-blast TEXT Optional file name for saving result of BLAST search
--sel-key TEXT Selection key to filter BLAST output.
Provide either a keyword, or 'host: <species>'
--plot-width INTEGER Width for the multi-alignment plot.
--blast-json FILE Path to a json file containing parameters for the blast search.
--email TEXT Email for Entrez search.
--multimer Store the output sequences for a multimer ColabFold run (from identical chains).
If not set, "--n-chains" will not be used.
--n-chains INTEGER Number of repeated chains that will be saved in csv file.
Requires calling the "--multimer" option first.
--gen-ref-pdb Fetch PDB of crystal structure for reference target, from the RCSB database.
--plot-width INTEGER Width for the multi-alignment plot.
--color-seq-match Color aminoacid matches in html alignment:
Red for exact match and yellow for same-
group match.
--align-start-idx INTEGER Start index for reference aminoacids in html
alignment (Useful when matching idxs to
PyMOL labels)
--max-mismatches INTEGER Maximum number of aminoacid group
missmatches to be allowed in color-seq-match
mode.
--custom-order TEXT Custom order of aligned sequences (not
including ref) can be provided as a string
with comma-sep indexes.
--loglevel [DEBUG|INFO|WARNING|ERROR|CRITICAL]
The log level to use. [default: INFO]
--help Show this message and exit.
The following optional arguments control the BLAST search:
The number of alignments is controled through the
--nalignoption and is set to 1000 by default. Other BLAST API parameters such as the number of hits and the number of descriptions are calculated internally to match the number given for--nalign.The expect value cutoff is controlled via
--e-thr, which is 10 by default.The blast output is saved to a CSV file, with the path specified via the
--save-blastoption.
The selection string has the same format as the previous tutorial, allowing both keyword and Entrez selections, via the --sel-key argument. Make sure to include the email in--email when requesting a an Entrez selection. The CLI will throw an error if this is not provided.
For the test case of a single protein chain of SARS-CoV-2 MPro, which we previously saved as sars-cov2-mpro.fasta, we run the following command:
# Starting from a .fasta file
drugforge-spectrum seq-alignment -f sars-cov2-mpro.fasta -t fasta \
--output-dir tutorial_results \
--sel-key "host: Homo sapiens OR organism: human" \
--email your.email@email.com \
--color-seq-match
# Starting from a .pdb file
drugforge-spectrum seq-alignment -f 8ya5.pdb -t pdb \
--output-dir tutorial_results \
--sel-key "host: Homo sapiens OR organism: human" \
--email your.email@email.com \
--color-seq-match
# Starting from a pre-computed BLAST search (.xml file)
drugforge-spectrum seq-alignment -f results.xml -t pre-calc \
--output-dir tutorial_results \
--sel-key "host: Homo sapiens OR organism: human" \
--email your.email@email.com \
--color-seq-match
All of the output files generated are named according to the title of the sequence given in the input fasta file, with an additional prefix that can be added through the --aln-output option if desired. The following files are saved in --results-folder:
A CSV file with the results from the BLAST search on the reference sequence (default path is
blast.csv)A
.fastafile with the ID, description and sequence of the sequences selected via the provided keyword.a CSV file with the ID and sequence of the sequences selected via the provided keyword, required for the structure alignment pipeline.
An
.htmlfile with a colored representation of the multi-sequence alignment. The width of the plot can be adjusted via--plot-width.A
.fastafile with the aligned sequences (with “-” representing gaps inserted)If the
--gen-ref-pdbflag is used, a PDB file corresponding to the crystal structure of the reference sequence. The RCSB entry with the highest resolution is retrieved.
The default setting to generate a ColabFold run CSV input is for monomers, but this can be adjusted for folding multimers. For example, starting from the fasta input file:
# Starting from a .fasta file, generate ColabFold input for a dimer fold
drugforge-spectrum seq-alignment -f sars-cov2-mpro.fasta -t fasta \
--output-dir tutorial_results \
--sel-key "host: Homo sapiens OR organism: human" \
--email your.email@email.com \
--multimer --nchains 2
The above will generate the necessary input for folding the related protein sequences as dimers.
Finally, the user can fetch a PDB crystal structures of the reference by optionally providing the flag --gen-ref-pdb. Note that this functionality requires previous installation of the ProDy module in the asapdiscovery environment.
Structure alignment usage
Based on a ColabFold run, as described in the tutorial, the struct-alignment command aligns the PDBs of the folded sequences listed in a .csv file and generates a PyMOL session for visualization.
The drugforge-spectrum struct-alignment CLI takes as input the csv file, generated from the seq-alignment run, the PDB file of the protein that we used as a reference for the two prior steps, the path to the folder where the results from the ColabFold run are stored (the $out_dir environment variable assigned to the run on the folding step), and the path to the directory where alignment results will be stored. Below is a list of these options, which can also be accessed via the
drugforge-spectrum struct-alignment --help option:
-f, --seq-file FILE File containing reference sequences
--pdb-file FILE Path to a pdb file containing a structure
--output-dir DIRECTORY The directory to output results to.
--cfold-results DIRECTORY Path to folder where all ColabFold results are stored.
--pdb-align TEXT Path to PDB to align. Not needed when
--cfold-results is given.
--struct-dir DIRECTORY Path to folder where structures to align are
stored. Not needed when --cfold-results or
--pdb-align is given.
--pymol-save TEXT Path to save pymol session with aligned
proteins.
--chain TEXT Chains to display on visualization ('A', 'B'
or 'both'). The default 'both' will align
wrt chain A but display both chains.
--color-by-rmsd Option to generate a PyMOL session were
targets are colored by RMSD with respect to
ref.
--cf-format TEXT Model used with ColabFold. Either
'alphafold2_ptm' or 'alphafold2_multimer_v3'
--loglevel [DEBUG|INFO|WARNING|ERROR|CRITICAL]
The log level to use. [default: INFO]
--help Show this message and exit.
Optionally, a custom name for the PyMOL session can be provided, with the default being aligned_proteins.pse. The format of the PDB file of the folded protein that ColabFold generates can also be controlled by the argument --cf-format. The default format is ``”alphafold2_ptm”``, which corresponds to a run using the alphafold2_ptm model and no relaxation step. If a multimer model was used for ColabFold, ``”alphafold2_multimer_v3”`` would be the correct format, although the string
can be verified by looking at the contents of the --cfold-results directory.
The following command will create a PyMOL session based on the ColabFold results stored in the directory sars_cf_results/:
# From the csv file and ColabFold files generated in the previous tutorial steps
drugforge-spectrum struct-alignment -f sars_tutorial_human_host.csv \
--pdb-file 8dz0.pdb \
--output-dir aligned_sars_tutorial \
--cfold-results sars_cf_results \
--cf-format alphafold2_multimer_v3 \
--chain "A" \
--color-by-rmsd
It is also possible to align protein complexes already stored on a directory (as PDB files), or from a single PDB file:
# From a single PDB file
drugforge-spectrum struct-alignment -f sars_tutorial_human_host.csv \
--pdb-file 8dz0.pdb \
--output-dir "aligned_sars_redo" \
--pdb-align "aligned_sars_tutorial/YP_009725295_1_aligned.pdb" \
--chain A \
--color-by-rmsd \
--pymol-save aligned_single_pdb.pse
# From the csv file and ColabFold files generated in the previous tutorial steps (dimer run)
drugforge-spectrum struct-alignment -f sars_tutorial_human_host.csv \
--pdb-file 8dz0.pdb \
--output-dir "aligned_sars_redo" \
--struct-dir "aligned_sars_tutorial" \
--chain A \
--color-by-rmsd \
--pymol-save aligned_structure_file.pse
Scoring usage
The CLI can be used to score a protein-ligand complex, or a directory with multiple structures. Different options can be given, that will determine what scoring methods will be used. By default, only the ChemGauss score and ligand pose RMSD are calculated, while the Binding pocket RMSD, AutoDock Vina score (works only if ADFR software suite and Meeko have been installed, see Requirements), and Gnina score (if
installed), can be requested as well. Running this call generate a csv file containing all the requested scores. --docking-csv provides the option to add the docking score from a previous drugforge-docking run, by providing the docking_scores_filtered_sorted.csv file generated from that run, but can take an empty string (" ", noting the space) as input if that feature is not desired.
The csv file with the scores will be have a column with the protein and/or ligand ID identifiers, which are determined based on the file names of the structures in --docking-dir. The defaults are set to work with the identifiers returned by BLAST in the sequence alignment tutorial (e.g., “YP_009725295”, which is SARS-CoV-2), but they can be specified with --protein-regex, which takes a regex pattern string. For example, --protein-regex '^([A-Za-z0-9\-]+)' will extract the target
name from MERS-CoV_ensitrelvir_folded.pdb, and --ligand-regex '^[^_]+_([^_]+)' extracts the ligand (ensitrelvir).
Note that the --ml-score flag requests scoring through ML models implemented within drugforge-ml, but this functionality is currenlty unavailable.
Options:
-d, --docking-dir PATH Path to directory where docked structures
are stored.
-f, --pdb_ref PATH Path to directory/file where crystal
structures are stored.
-o, --out-dir TEXT Path to directory where scoring results will
be stored.
--docking-csv PATH Path to csv files with docking results.
--target [EV-D68-Capsid|SARS-CoV-2-N-protein|EV-A71-3Cpro|EV-A71-2Apro|EV-A71-Capsid|DENV-NS2B-NS3pro|SARS-CoV-2-Mpro|EV-D68-3Cpro|ZIKV-RdRppro|SARS-CoV-2-Mac1|MERS-CoV-Mpro|ZIKV-NS2B-NS3pro]
The target for the workflow [required]
--vina-score Whether to run vina scoring.
--vina-box-x FLOAT coordinate x of vina box.
--vina-box-y FLOAT coordinate y of vina box.
--vina-box-z FLOAT coordinate z of vina box.
--path-to-grid-prep PATH Path to .py file that calculates grid for
Vina.
--docking-vina Whether to run docking on vina.
--ligand-regex TEXT Pattern for extracting ligand ID from file
name.
--protein-regex TEXT Pattern for extracting protein ID from file
name.
--minimize Whether to minimize the pdb structures
before running scoring.
--md-openmm-platform TEXT The OpenMM platform to use for MD
minimization.
[CPU|CUDA|OpenCL|Reference|Fastest]
--ml-score Whether to employ implemented ML models to
score poses.
--bsite-rmsd Whether to calculate binding site RMSD (only
relevant when the ref pdb is the same target
as the docked complex).
--dock-chain TEXT Chain ID of main chain in docked complex
pdb(s).
--ref-chain TEXT Chain ID of main chain in reference pdb(s).
--lig-resname TEXT Residue name of ligand in reference pdb(s).
--gnina-score Whether to run gnina scoring.
--gnina-script TEXT Path to bash script that runs Gnina CLI.
--gnina-out-dir PATH Directory for gnina output.
--log-level TEXT Logging level.
--input-json FILE Path to a json file containing the inputs to
the workflow, WARNING: overrides all other
inputs.
--help Show this message and exit.
The following command will run the scoring workflow without AutoDock Vina, assuming MERS-CoV_Lig-ensitrelvir_folded.pdb is saved on a folder docked_folder, with other complexes like SARS-CoV-2_Lig-ensitrelvir_folded.pdb, or SARS-CoV-2_Lig-someligand_folded.pdb:
drugforge-spectrum score -d docked_folder/ \
-f 8dz0.pdb \
-o score_results/ \
--docking-csv " " \
--target SARS-CoV-2-Mpro \
--dock-chain "1" \
--ref-chain "A" \
--bsite-rmsd \
--protein-regex '^([A-Za-z0-9\\-]+)' \
--ligand-regex 'Lig-[^_]+'
Note that --dock-chain and --ref-chain need to correspond to the respective pdb files. The run will throw an error if it can’t find a chain with that identifier in the PDB file. To run Vina scoring, if all requirements are installed, with the center of the ligand box specified via the flag --vina-box-[x,y,z]:
drugforge-spectrum score -d docked_folder/ \
-f 8dz0.pdb \
-o score_results/ \
--docking-csv " " \
--target SARS-CoV-2-Mpro \
--dock-chain "1" \
--ref-chain "A" \
--bsite-rmsd \
--protein-regex '^([A-Za-z0-9\\-]+)' \
--ligand-regex 'Lig-[^_]+' \
--vina-score \
--vina-box-x -22.0 \
--vina-box-y 6.0 \
--vina-box-z 25.0